{"title":"B36 硼吩納米薄片上溴丙酮吸附行為的計算研究","authors":"Meriem Taier, Hamza Allal, Salim Bousba, Fathi Bouhadiouche, Soumeya Maza, Maamar Damous, Ahlem Boussadia","doi":"10.1007/s10825-024-02192-3","DOIUrl":null,"url":null,"abstract":"<div><p>Density functional theory (DFT) methods are employed to investigate the capability of B<sub>36</sub> borophene nanosheets as sensors for detecting the bromoacetone (BCT) molecule. An evaluation of the structural and electronic properties of both BCT and B<sub>36</sub> borophene is conducted. Subsequently, through computed metrics such as adsorption energy, charge density difference, and density of states, the interaction between B<sub>36</sub> and the BCT molecule is examined via dispersion-corrected density functional theory (DFT). Employing the reduced density gradient approach for the analysis of non-covalent interactions, we further explored the nature of these interactions. The obtained results illustrate that B<sub>36</sub> borophene nanosheets serve as effective sensors for the BCT molecule, showcasing their ability to adsorb up to five BCT molecules through an exothermic process. BCT molecules chemiadsorb onto B<sub>36</sub> borophene by forming B‒O covalent bonds, engaging the oxygen atom of the carbonyl group in BCT with the edge boron atoms of B<sub>36</sub> borophene. Additionally, BCT molecules physio-adsorb on both the concave and convex sides of B<sub>36</sub> borophene, facilitated by van der Waals interactions. Ab-initio molecular dynamic simulations confirm the thermal stability of the BCT@B<sub>36</sub> concave and convex complexes at both 300 K and 400 K.</p></div>","PeriodicalId":620,"journal":{"name":"Journal of Computational Electronics","volume":"23 5","pages":"931 - 944"},"PeriodicalIF":2.8000,"publicationDate":"2024-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A computational investigation on the adsorption behavior of bromoacetone on B36 borophene nanosheets\",\"authors\":\"Meriem Taier, Hamza Allal, Salim Bousba, Fathi Bouhadiouche, Soumeya Maza, Maamar Damous, Ahlem Boussadia\",\"doi\":\"10.1007/s10825-024-02192-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Density functional theory (DFT) methods are employed to investigate the capability of B<sub>36</sub> borophene nanosheets as sensors for detecting the bromoacetone (BCT) molecule. An evaluation of the structural and electronic properties of both BCT and B<sub>36</sub> borophene is conducted. Subsequently, through computed metrics such as adsorption energy, charge density difference, and density of states, the interaction between B<sub>36</sub> and the BCT molecule is examined via dispersion-corrected density functional theory (DFT). Employing the reduced density gradient approach for the analysis of non-covalent interactions, we further explored the nature of these interactions. The obtained results illustrate that B<sub>36</sub> borophene nanosheets serve as effective sensors for the BCT molecule, showcasing their ability to adsorb up to five BCT molecules through an exothermic process. BCT molecules chemiadsorb onto B<sub>36</sub> borophene by forming B‒O covalent bonds, engaging the oxygen atom of the carbonyl group in BCT with the edge boron atoms of B<sub>36</sub> borophene. Additionally, BCT molecules physio-adsorb on both the concave and convex sides of B<sub>36</sub> borophene, facilitated by van der Waals interactions. Ab-initio molecular dynamic simulations confirm the thermal stability of the BCT@B<sub>36</sub> concave and convex complexes at both 300 K and 400 K.</p></div>\",\"PeriodicalId\":620,\"journal\":{\"name\":\"Journal of Computational Electronics\",\"volume\":\"23 5\",\"pages\":\"931 - 944\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-06-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Electronics\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10825-024-02192-3\",\"RegionNum\":4,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ENGINEERING, ELECTRICAL & ELECTRONIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Electronics","FirstCategoryId":"5","ListUrlMain":"https://link.springer.com/article/10.1007/s10825-024-02192-3","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENGINEERING, ELECTRICAL & ELECTRONIC","Score":null,"Total":0}
A computational investigation on the adsorption behavior of bromoacetone on B36 borophene nanosheets
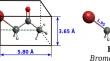
Density functional theory (DFT) methods are employed to investigate the capability of B36 borophene nanosheets as sensors for detecting the bromoacetone (BCT) molecule. An evaluation of the structural and electronic properties of both BCT and B36 borophene is conducted. Subsequently, through computed metrics such as adsorption energy, charge density difference, and density of states, the interaction between B36 and the BCT molecule is examined via dispersion-corrected density functional theory (DFT). Employing the reduced density gradient approach for the analysis of non-covalent interactions, we further explored the nature of these interactions. The obtained results illustrate that B36 borophene nanosheets serve as effective sensors for the BCT molecule, showcasing their ability to adsorb up to five BCT molecules through an exothermic process. BCT molecules chemiadsorb onto B36 borophene by forming B‒O covalent bonds, engaging the oxygen atom of the carbonyl group in BCT with the edge boron atoms of B36 borophene. Additionally, BCT molecules physio-adsorb on both the concave and convex sides of B36 borophene, facilitated by van der Waals interactions. Ab-initio molecular dynamic simulations confirm the thermal stability of the BCT@B36 concave and convex complexes at both 300 K and 400 K.
期刊介绍:
he Journal of Computational Electronics brings together research on all aspects of modeling and simulation of modern electronics. This includes optical, electronic, mechanical, and quantum mechanical aspects, as well as research on the underlying mathematical algorithms and computational details. The related areas of energy conversion/storage and of molecular and biological systems, in which the thrust is on the charge transport, electronic, mechanical, and optical properties, are also covered.
In particular, we encourage manuscripts dealing with device simulation; with optical and optoelectronic systems and photonics; with energy storage (e.g. batteries, fuel cells) and harvesting (e.g. photovoltaic), with simulation of circuits, VLSI layout, logic and architecture (based on, for example, CMOS devices, quantum-cellular automata, QBITs, or single-electron transistors); with electromagnetic simulations (such as microwave electronics and components); or with molecular and biological systems. However, in all these cases, the submitted manuscripts should explicitly address the electronic properties of the relevant systems, materials, or devices and/or present novel contributions to the physical models, computational strategies, or numerical algorithms.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
