{"title":"通过网格投影原子指纹的卷积网络学习自洽电子密度","authors":"Ryong-Gyu Lee, Yong-Hoon Kim","doi":"10.1038/s41524-024-01433-0","DOIUrl":null,"url":null,"abstract":"<p>The self-consistent field (SCF) generation of the three-dimensional (3D) electron density distribution (<i>ρ</i>) represents a fundamental aspect of density functional theory (DFT) and related first-principles calculations, and how one can shorten or bypass the SCF loop represents a critical question in electronic structure theory from both practical and fundamental standpoints. Herein, a machine learning strategy, DeepSCF, is presented in which the map between the SCF <i>ρ</i> and the initial guess density (<i>ρ</i><sub>0</sub>) constructed by the summation of neutral atomic densities is learned using 3D convolutional neural networks (CNNs). High accuracy and transferability of DeepSCF are achieved by first encoding <i>ρ</i><sub>0</sub> on a 3D grid and then expanding the input features to include atomic fingerprints beyond <i>ρ</i><sub>0</sub>. The prediction of the residual density (δ<i>ρ</i>) rather than <i>ρ</i> itself is targeted, and given that δ<i>ρ</i> is indicative of chemical bonding information, a dataset of small-sized organic molecules featuring diverse bonding characters is adopted. The fidelity of DeepSCF is finally enhanced by subjecting the atomic geometries of the dataset to random rotations and strains. The effectiveness of DeepSCF is demonstrated using a complex carbon nanotube-based DNA sequencer model. This work evidences that the nearsightedness in electronic structure can be optimally represented via the spatial locality in CNNs, offering insight into the success of various machine learning-based atomistic materials simulations.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"30 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-10-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints\",\"authors\":\"Ryong-Gyu Lee, Yong-Hoon Kim\",\"doi\":\"10.1038/s41524-024-01433-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The self-consistent field (SCF) generation of the three-dimensional (3D) electron density distribution (<i>ρ</i>) represents a fundamental aspect of density functional theory (DFT) and related first-principles calculations, and how one can shorten or bypass the SCF loop represents a critical question in electronic structure theory from both practical and fundamental standpoints. Herein, a machine learning strategy, DeepSCF, is presented in which the map between the SCF <i>ρ</i> and the initial guess density (<i>ρ</i><sub>0</sub>) constructed by the summation of neutral atomic densities is learned using 3D convolutional neural networks (CNNs). High accuracy and transferability of DeepSCF are achieved by first encoding <i>ρ</i><sub>0</sub> on a 3D grid and then expanding the input features to include atomic fingerprints beyond <i>ρ</i><sub>0</sub>. The prediction of the residual density (δ<i>ρ</i>) rather than <i>ρ</i> itself is targeted, and given that δ<i>ρ</i> is indicative of chemical bonding information, a dataset of small-sized organic molecules featuring diverse bonding characters is adopted. The fidelity of DeepSCF is finally enhanced by subjecting the atomic geometries of the dataset to random rotations and strains. The effectiveness of DeepSCF is demonstrated using a complex carbon nanotube-based DNA sequencer model. This work evidences that the nearsightedness in electronic structure can be optimally represented via the spatial locality in CNNs, offering insight into the success of various machine learning-based atomistic materials simulations.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"30 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-10-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01433-0\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01433-0","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints
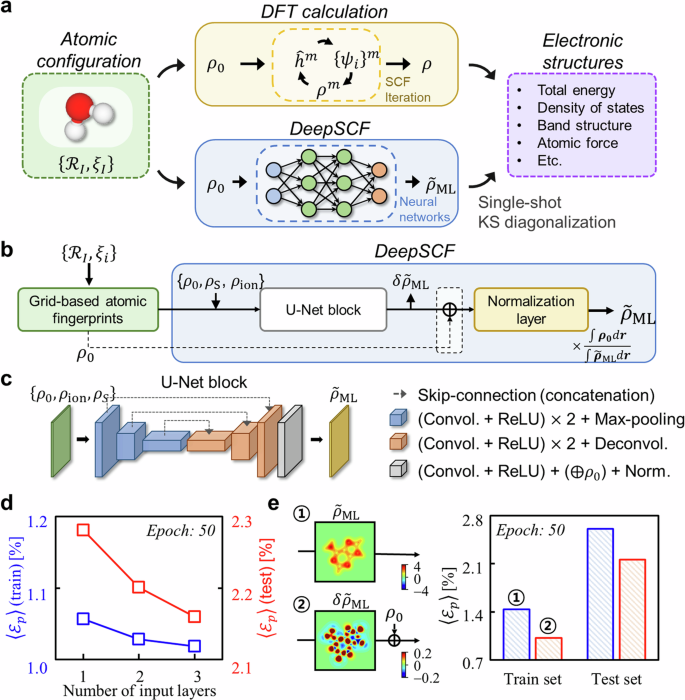
The self-consistent field (SCF) generation of the three-dimensional (3D) electron density distribution (ρ) represents a fundamental aspect of density functional theory (DFT) and related first-principles calculations, and how one can shorten or bypass the SCF loop represents a critical question in electronic structure theory from both practical and fundamental standpoints. Herein, a machine learning strategy, DeepSCF, is presented in which the map between the SCF ρ and the initial guess density (ρ0) constructed by the summation of neutral atomic densities is learned using 3D convolutional neural networks (CNNs). High accuracy and transferability of DeepSCF are achieved by first encoding ρ0 on a 3D grid and then expanding the input features to include atomic fingerprints beyond ρ0. The prediction of the residual density (δρ) rather than ρ itself is targeted, and given that δρ is indicative of chemical bonding information, a dataset of small-sized organic molecules featuring diverse bonding characters is adopted. The fidelity of DeepSCF is finally enhanced by subjecting the atomic geometries of the dataset to random rotations and strains. The effectiveness of DeepSCF is demonstrated using a complex carbon nanotube-based DNA sequencer model. This work evidences that the nearsightedness in electronic structure can be optimally represented via the spatial locality in CNNs, offering insight into the success of various machine learning-based atomistic materials simulations.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
