Ebert Alvares, Kai Sellschopp, Bo Wang, ShinYoung Kang, Thomas Klassen, Brandon C. Wood, Tae Wook Heo, Paul Jerabek, Claudio Pistidda
{"title":"金属氢化物相间的多尺度建模--解耦化学机械能的量化","authors":"Ebert Alvares, Kai Sellschopp, Bo Wang, ShinYoung Kang, Thomas Klassen, Brandon C. Wood, Tae Wook Heo, Paul Jerabek, Claudio Pistidda","doi":"10.1038/s41524-024-01424-1","DOIUrl":null,"url":null,"abstract":"<p>The quantification of interphase properties between metals and their corresponding hydrides is crucial for modeling the thermodynamics and kinetics of the hydrogenation processes in solid-state hydrogen storage materials. In particular, interphase boundary energies assume a pivotal role in determining the kinetics of nucleation, growth, and coarsening of hydrides, alongside accompanying morphological evolution during hydrogenation. The total interphase energy arises from both chemical bonding and mechanical strains in these solid-state systems. Since these contributions are usually coupled, it is challenging to distinguish via conventional computational approaches. Here, a comprehensive atomistic modeling methodology is developed to decouple chemical and mechanical energy contributions using first-principles calculations, of which feasibility is demonstrated by quantifying chemical and elastic strain energies of key interfaces within the FeTi metal-hydride system. Derived materials parameters are then employed for mesoscopic micromechanical analysis, predicting crystallographic orientations in line with experimental observations. The multiscale approach outlined verifies the importance of the chemo-mechanical interplay in the morphological evolution of growing hydride phases, and can be generalized to investigate other systems. In addition, it can streamline the design of atomistic models for the quantitative evaluation of interphase properties between dissimilar phases and allow for efficient predictions of their preferred phase boundary orientations.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"212 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-10-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Multiscale modeling of metal-hydride interphases—quantification of decoupled chemo-mechanical energies\",\"authors\":\"Ebert Alvares, Kai Sellschopp, Bo Wang, ShinYoung Kang, Thomas Klassen, Brandon C. Wood, Tae Wook Heo, Paul Jerabek, Claudio Pistidda\",\"doi\":\"10.1038/s41524-024-01424-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The quantification of interphase properties between metals and their corresponding hydrides is crucial for modeling the thermodynamics and kinetics of the hydrogenation processes in solid-state hydrogen storage materials. In particular, interphase boundary energies assume a pivotal role in determining the kinetics of nucleation, growth, and coarsening of hydrides, alongside accompanying morphological evolution during hydrogenation. The total interphase energy arises from both chemical bonding and mechanical strains in these solid-state systems. Since these contributions are usually coupled, it is challenging to distinguish via conventional computational approaches. Here, a comprehensive atomistic modeling methodology is developed to decouple chemical and mechanical energy contributions using first-principles calculations, of which feasibility is demonstrated by quantifying chemical and elastic strain energies of key interfaces within the FeTi metal-hydride system. Derived materials parameters are then employed for mesoscopic micromechanical analysis, predicting crystallographic orientations in line with experimental observations. The multiscale approach outlined verifies the importance of the chemo-mechanical interplay in the morphological evolution of growing hydride phases, and can be generalized to investigate other systems. In addition, it can streamline the design of atomistic models for the quantitative evaluation of interphase properties between dissimilar phases and allow for efficient predictions of their preferred phase boundary orientations.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"212 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-10-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01424-1\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01424-1","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Multiscale modeling of metal-hydride interphases—quantification of decoupled chemo-mechanical energies
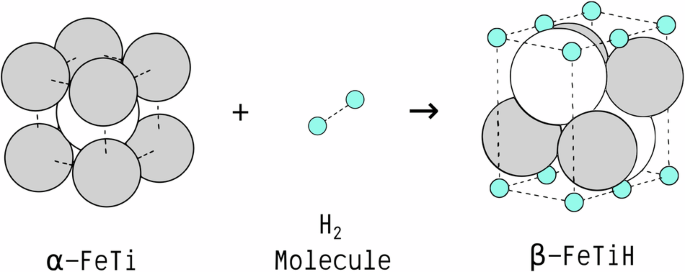
The quantification of interphase properties between metals and their corresponding hydrides is crucial for modeling the thermodynamics and kinetics of the hydrogenation processes in solid-state hydrogen storage materials. In particular, interphase boundary energies assume a pivotal role in determining the kinetics of nucleation, growth, and coarsening of hydrides, alongside accompanying morphological evolution during hydrogenation. The total interphase energy arises from both chemical bonding and mechanical strains in these solid-state systems. Since these contributions are usually coupled, it is challenging to distinguish via conventional computational approaches. Here, a comprehensive atomistic modeling methodology is developed to decouple chemical and mechanical energy contributions using first-principles calculations, of which feasibility is demonstrated by quantifying chemical and elastic strain energies of key interfaces within the FeTi metal-hydride system. Derived materials parameters are then employed for mesoscopic micromechanical analysis, predicting crystallographic orientations in line with experimental observations. The multiscale approach outlined verifies the importance of the chemo-mechanical interplay in the morphological evolution of growing hydride phases, and can be generalized to investigate other systems. In addition, it can streamline the design of atomistic models for the quantitative evaluation of interphase properties between dissimilar phases and allow for efficient predictions of their preferred phase boundary orientations.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
