Kazuma Ito, Tatsuya Yokoi, Katsutoshi Hyodo, Hideki Mori
{"title":"针对Œ±-Fe 中一般晶界的具有 DFT 精确度的机器学习原子间势","authors":"Kazuma Ito, Tatsuya Yokoi, Katsutoshi Hyodo, Hideki Mori","doi":"10.1038/s41524-024-01451-y","DOIUrl":null,"url":null,"abstract":"<p>To advance the development of high-strength polycrystalline metallic materials towards achieving carbon neutrality, it is essential to design materials in which the atomic level control of general grain boundaries (GGBs), which govern the material properties, is achieved. However, owing to the complex and diverse structures of GGBs, there have been no reports on interatomic potentials capable of reproducing them. This accuracy is essential for conducting molecular dynamics analyses to derive material design guidelines. In this study, we constructed a machine learning interatomic potential (MLIP) with density functional theory (DFT) accuracy to model the energy, atomic structure, and dynamics of arbitrary grain boundaries (GBs), including GGBs, in Œ±-Fe. Specifically, we employed a training dataset comprising diverse atomic structures generated based on crystal space groups. The GGB accuracy was evaluated by directly comparing with DFT calculations performed on cells cut near GBs from nano-polycrystals, and extrapolation grades of the local atomic environment based on active learning methods for the entire nano-polycrystal. Furthermore, we analyzed the GB energy and atomic structure in Œ±-Fe polycrystals through large-scale molecular dynamics analysis using the constructed MLIP. The average GB energy of Œ±-Fe polycrystals calculated by the constructed MLIP is 1.57‚ÄâJ/m<sup>2</sup>, exhibiting good agreement with experimental predictions. Our findings demonstrate the methodology for constructing an MLIP capable of representing GGBs with high accuracy, thereby paving the way for materials design based on computational materials science for polycrystalline materials.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"232 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine learning interatomic potential with DFT accuracy for general grain boundaries in Œ±-Fe\",\"authors\":\"Kazuma Ito, Tatsuya Yokoi, Katsutoshi Hyodo, Hideki Mori\",\"doi\":\"10.1038/s41524-024-01451-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>To advance the development of high-strength polycrystalline metallic materials towards achieving carbon neutrality, it is essential to design materials in which the atomic level control of general grain boundaries (GGBs), which govern the material properties, is achieved. However, owing to the complex and diverse structures of GGBs, there have been no reports on interatomic potentials capable of reproducing them. This accuracy is essential for conducting molecular dynamics analyses to derive material design guidelines. In this study, we constructed a machine learning interatomic potential (MLIP) with density functional theory (DFT) accuracy to model the energy, atomic structure, and dynamics of arbitrary grain boundaries (GBs), including GGBs, in Œ±-Fe. Specifically, we employed a training dataset comprising diverse atomic structures generated based on crystal space groups. The GGB accuracy was evaluated by directly comparing with DFT calculations performed on cells cut near GBs from nano-polycrystals, and extrapolation grades of the local atomic environment based on active learning methods for the entire nano-polycrystal. Furthermore, we analyzed the GB energy and atomic structure in Œ±-Fe polycrystals through large-scale molecular dynamics analysis using the constructed MLIP. The average GB energy of Œ±-Fe polycrystals calculated by the constructed MLIP is 1.57‚ÄâJ/m<sup>2</sup>, exhibiting good agreement with experimental predictions. Our findings demonstrate the methodology for constructing an MLIP capable of representing GGBs with high accuracy, thereby paving the way for materials design based on computational materials science for polycrystalline materials.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"232 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-11-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01451-y\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01451-y","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Machine learning interatomic potential with DFT accuracy for general grain boundaries in α-Fe
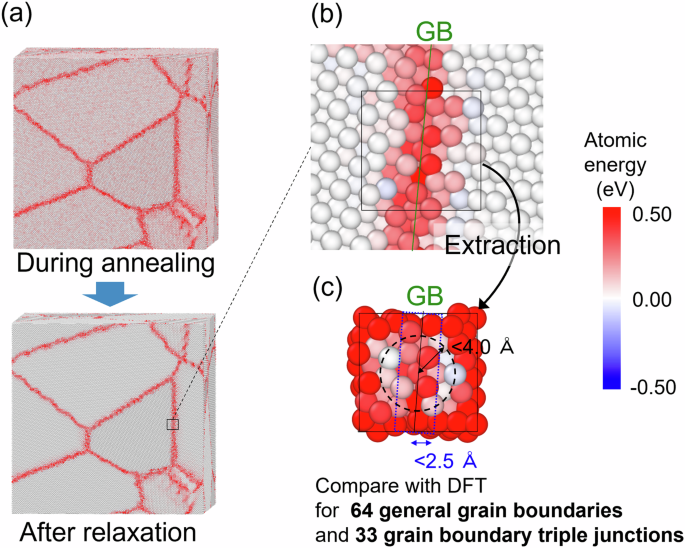
To advance the development of high-strength polycrystalline metallic materials towards achieving carbon neutrality, it is essential to design materials in which the atomic level control of general grain boundaries (GGBs), which govern the material properties, is achieved. However, owing to the complex and diverse structures of GGBs, there have been no reports on interatomic potentials capable of reproducing them. This accuracy is essential for conducting molecular dynamics analyses to derive material design guidelines. In this study, we constructed a machine learning interatomic potential (MLIP) with density functional theory (DFT) accuracy to model the energy, atomic structure, and dynamics of arbitrary grain boundaries (GBs), including GGBs, in α-Fe. Specifically, we employed a training dataset comprising diverse atomic structures generated based on crystal space groups. The GGB accuracy was evaluated by directly comparing with DFT calculations performed on cells cut near GBs from nano-polycrystals, and extrapolation grades of the local atomic environment based on active learning methods for the entire nano-polycrystal. Furthermore, we analyzed the GB energy and atomic structure in α-Fe polycrystals through large-scale molecular dynamics analysis using the constructed MLIP. The average GB energy of α-Fe polycrystals calculated by the constructed MLIP is 1.57 J/m2, exhibiting good agreement with experimental predictions. Our findings demonstrate the methodology for constructing an MLIP capable of representing GGBs with high accuracy, thereby paving the way for materials design based on computational materials science for polycrystalline materials.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
