Rodrigo Arcoverde Cerveira , Klara Lenart , Marcel Martin , Matthew James Hinchcliff , Fredrika Hellgren , Kewei Ye , Juliana Assis Geraldo , Taras Kreslavsky , Sebastian Ols , Karin Loré
{"title":"Scifer:用于大规模整合桑格测序和流式细胞仪指数分选单细胞数据的 R/Bioconductor 软件包","authors":"Rodrigo Arcoverde Cerveira , Klara Lenart , Marcel Martin , Matthew James Hinchcliff , Fredrika Hellgren , Kewei Ye , Juliana Assis Geraldo , Taras Kreslavsky , Sebastian Ols , Karin Loré","doi":"10.1016/j.immuno.2024.100046","DOIUrl":null,"url":null,"abstract":"<div><div>Sanger sequencing remains widely used in various experimental contexts, often in combination with flow cytometry for indexing specific cell populations. However, existing software lacks the capability to automate quality control (QC) of raw Sanger sequencing data and integrate it with flow cytometry information on a large scale. Here, we introduce scifer, an R package now available in the latest release of Bioconductor (3.20) showcasing its effectiveness in seamlessly integrating these types of data as demonstrated by analyses of B cell and T cell receptor sequences. Scifer preprocesses raw data from index sorts and immune receptor Sanger sequencing. It identifies high-quality sequences based on selected parameters, such as length, Phred scores, and heavy-chain complementarity-determining region 3 (HCDR3) quality. As a result, the quality of germline assignments is significantly increased and spurious variable gene mutations are reduced. Scifer is automated and can process thousands of sequences in less than an hour. Its output provides quality control reports, FASTA files, summarized tables, and electropherograms for manual inspection. In summary, scifer is a user-friendly software that speeds up the analysis of immune receptor repertoire sequences, offering wide applicability.</div></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"16 ","pages":"Article 100046"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Scifer: An R/Bioconductor package for large-scale integration of Sanger sequencing and flow cytometry data of index-sorted single cells\",\"authors\":\"Rodrigo Arcoverde Cerveira , Klara Lenart , Marcel Martin , Matthew James Hinchcliff , Fredrika Hellgren , Kewei Ye , Juliana Assis Geraldo , Taras Kreslavsky , Sebastian Ols , Karin Loré\",\"doi\":\"10.1016/j.immuno.2024.100046\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Sanger sequencing remains widely used in various experimental contexts, often in combination with flow cytometry for indexing specific cell populations. However, existing software lacks the capability to automate quality control (QC) of raw Sanger sequencing data and integrate it with flow cytometry information on a large scale. Here, we introduce scifer, an R package now available in the latest release of Bioconductor (3.20) showcasing its effectiveness in seamlessly integrating these types of data as demonstrated by analyses of B cell and T cell receptor sequences. Scifer preprocesses raw data from index sorts and immune receptor Sanger sequencing. It identifies high-quality sequences based on selected parameters, such as length, Phred scores, and heavy-chain complementarity-determining region 3 (HCDR3) quality. As a result, the quality of germline assignments is significantly increased and spurious variable gene mutations are reduced. Scifer is automated and can process thousands of sequences in less than an hour. Its output provides quality control reports, FASTA files, summarized tables, and electropherograms for manual inspection. In summary, scifer is a user-friendly software that speeds up the analysis of immune receptor repertoire sequences, offering wide applicability.</div></div>\",\"PeriodicalId\":73343,\"journal\":{\"name\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"volume\":\"16 \",\"pages\":\"Article 100046\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2667119024000168\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119024000168","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/29 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
桑格测序仍被广泛应用于各种实验中,通常与流式细胞仪结合使用,对特定细胞群进行索引。然而,现有软件缺乏对原始 Sanger 测序数据进行自动质量控制(QC)并将其与流式细胞仪信息大规模整合的能力。在这里,我们将介绍 scifer,这是一个 R 软件包,目前可在最新发布的 Bioconductor 3.20 中使用,通过对 B 细胞和 T 细胞受体序列的分析,我们展示了它在无缝整合这些类型数据方面的有效性。Scifer 对来自索引分类和免疫受体 Sanger 测序的原始数据进行预处理。它根据长度、Phred 分数和重链互补决定区 3 (HCDR3) 质量等选定参数识别高质量序列。因此,种系分配的质量大大提高,虚假的可变基因突变也减少了。Scifer 是自动化的,可在一小时内处理数千条序列。其输出结果包括质量控制报告、FASTA 文件、汇总表和供人工检查的电图。总之,scifer 是一款用户友好型软件,可加快免疫受体序列的分析速度,具有广泛的适用性。
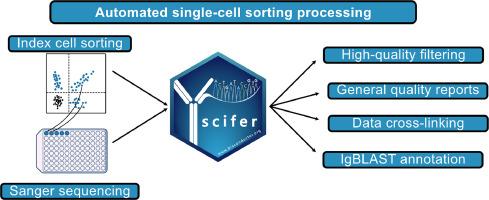
Scifer: An R/Bioconductor package for large-scale integration of Sanger sequencing and flow cytometry data of index-sorted single cells
Sanger sequencing remains widely used in various experimental contexts, often in combination with flow cytometry for indexing specific cell populations. However, existing software lacks the capability to automate quality control (QC) of raw Sanger sequencing data and integrate it with flow cytometry information on a large scale. Here, we introduce scifer, an R package now available in the latest release of Bioconductor (3.20) showcasing its effectiveness in seamlessly integrating these types of data as demonstrated by analyses of B cell and T cell receptor sequences. Scifer preprocesses raw data from index sorts and immune receptor Sanger sequencing. It identifies high-quality sequences based on selected parameters, such as length, Phred scores, and heavy-chain complementarity-determining region 3 (HCDR3) quality. As a result, the quality of germline assignments is significantly increased and spurious variable gene mutations are reduced. Scifer is automated and can process thousands of sequences in less than an hour. Its output provides quality control reports, FASTA files, summarized tables, and electropherograms for manual inspection. In summary, scifer is a user-friendly software that speeds up the analysis of immune receptor repertoire sequences, offering wide applicability.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
