Leonardo Pereira de Araújo , Maria Eduarda Carvalho Dias , Gislaine Cristina Scodeler , Ana de Souza Santos , Letícia Martins Soares , Patrícia Paiva Corsetti , Ana Carolina Barbosa Padovan , Nelson José de Freitas Silveira , Leonardo Augusto de Almeida
{"title":"利用计算机方法鉴定SARS-CoV-2结构蛋白的表位,获得保守的合理免疫原性肽","authors":"Leonardo Pereira de Araújo , Maria Eduarda Carvalho Dias , Gislaine Cristina Scodeler , Ana de Souza Santos , Letícia Martins Soares , Patrícia Paiva Corsetti , Ana Carolina Barbosa Padovan , Nelson José de Freitas Silveira , Leonardo Augusto de Almeida","doi":"10.1016/j.immuno.2022.100015","DOIUrl":null,"url":null,"abstract":"<div><p>The short time between the first cases of COVID-19 and the declaration of a pandemic initiated the search for ways to stop the spread of SARS-CoV-2. There are great expectations regarding the development of effective vaccines that protect against all variants, and in the search for it, we hypothesized the obtention of a predicted rational immunogenic peptide from structural components of SARS-CoV-2 might help the vaccine research direction. In the search for a candidate of an immunogenic peptide of the SARS-CoV-2 envelope (E), membrane (M), nucleocapsid (N), or spike (S) proteins, we access the predicted sequences of each protein after the genome sequenced worldwide. We obtained the consensus amino acid sequences of about 14,441 sequences of each protein of each continent and the worldwide consensus sequence. For epitope identification and characterization from each consensus structural protein related to MHC-I or MHC-II interaction and B-cell receptor recognition, we used the IEDB reaching 68 epitopes to E, 174 to M, 245 to N, and 833 to S proteins. To select an epitope with the highest probability of binding to the MHC or BCR, all epitopes of each consensus sequence were aligned. The curation indicated 1, 4, 8, and 21 selected epitopes for E, M, N, and S proteins, respectively. Those epitopes were tested in silico for antigenicity obtaining 16 antigenic epitopes. Physicochemical properties and allergenicity evaluation of the obtained epitopes were done. Ranking the results, we obtained one epitope of each protein except for the S protein that presented two epitopes after the selection. To check the 3D position of each selected epitope in the protein structure, we used molecular homology modeling. Afterward, each selected epitope was evaluated by molecular docking to reference MHC-I or MHC-II allelic protein sequences. Taken together, the results obtained in this study showed a rational search for a putative immunogenic peptide of SARS-CoV-2 structural proteins that can improve vaccine development using in silico approaches. The epitopes selected represent the most conserved sequence of new coronavirus and may be used in a variety of vaccine development strategies since they are also presented in the described variants of SARS-CoV-2.</p></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"7 ","pages":"Article 100015"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9188263/pdf/","citationCount":"3","resultStr":"{\"title\":\"Epitope identification of SARS-CoV-2 structural proteins using in silico approaches to obtain a conserved rational immunogenic peptide\",\"authors\":\"Leonardo Pereira de Araújo , Maria Eduarda Carvalho Dias , Gislaine Cristina Scodeler , Ana de Souza Santos , Letícia Martins Soares , Patrícia Paiva Corsetti , Ana Carolina Barbosa Padovan , Nelson José de Freitas Silveira , Leonardo Augusto de Almeida\",\"doi\":\"10.1016/j.immuno.2022.100015\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The short time between the first cases of COVID-19 and the declaration of a pandemic initiated the search for ways to stop the spread of SARS-CoV-2. There are great expectations regarding the development of effective vaccines that protect against all variants, and in the search for it, we hypothesized the obtention of a predicted rational immunogenic peptide from structural components of SARS-CoV-2 might help the vaccine research direction. In the search for a candidate of an immunogenic peptide of the SARS-CoV-2 envelope (E), membrane (M), nucleocapsid (N), or spike (S) proteins, we access the predicted sequences of each protein after the genome sequenced worldwide. We obtained the consensus amino acid sequences of about 14,441 sequences of each protein of each continent and the worldwide consensus sequence. For epitope identification and characterization from each consensus structural protein related to MHC-I or MHC-II interaction and B-cell receptor recognition, we used the IEDB reaching 68 epitopes to E, 174 to M, 245 to N, and 833 to S proteins. To select an epitope with the highest probability of binding to the MHC or BCR, all epitopes of each consensus sequence were aligned. The curation indicated 1, 4, 8, and 21 selected epitopes for E, M, N, and S proteins, respectively. Those epitopes were tested in silico for antigenicity obtaining 16 antigenic epitopes. Physicochemical properties and allergenicity evaluation of the obtained epitopes were done. Ranking the results, we obtained one epitope of each protein except for the S protein that presented two epitopes after the selection. To check the 3D position of each selected epitope in the protein structure, we used molecular homology modeling. Afterward, each selected epitope was evaluated by molecular docking to reference MHC-I or MHC-II allelic protein sequences. Taken together, the results obtained in this study showed a rational search for a putative immunogenic peptide of SARS-CoV-2 structural proteins that can improve vaccine development using in silico approaches. The epitopes selected represent the most conserved sequence of new coronavirus and may be used in a variety of vaccine development strategies since they are also presented in the described variants of SARS-CoV-2.</p></div>\",\"PeriodicalId\":73343,\"journal\":{\"name\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"volume\":\"7 \",\"pages\":\"Article 100015\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9188263/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2667119022000076\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/6/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119022000076","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/11 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Epitope identification of SARS-CoV-2 structural proteins using in silico approaches to obtain a conserved rational immunogenic peptide
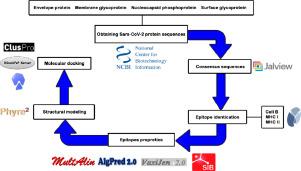
The short time between the first cases of COVID-19 and the declaration of a pandemic initiated the search for ways to stop the spread of SARS-CoV-2. There are great expectations regarding the development of effective vaccines that protect against all variants, and in the search for it, we hypothesized the obtention of a predicted rational immunogenic peptide from structural components of SARS-CoV-2 might help the vaccine research direction. In the search for a candidate of an immunogenic peptide of the SARS-CoV-2 envelope (E), membrane (M), nucleocapsid (N), or spike (S) proteins, we access the predicted sequences of each protein after the genome sequenced worldwide. We obtained the consensus amino acid sequences of about 14,441 sequences of each protein of each continent and the worldwide consensus sequence. For epitope identification and characterization from each consensus structural protein related to MHC-I or MHC-II interaction and B-cell receptor recognition, we used the IEDB reaching 68 epitopes to E, 174 to M, 245 to N, and 833 to S proteins. To select an epitope with the highest probability of binding to the MHC or BCR, all epitopes of each consensus sequence were aligned. The curation indicated 1, 4, 8, and 21 selected epitopes for E, M, N, and S proteins, respectively. Those epitopes were tested in silico for antigenicity obtaining 16 antigenic epitopes. Physicochemical properties and allergenicity evaluation of the obtained epitopes were done. Ranking the results, we obtained one epitope of each protein except for the S protein that presented two epitopes after the selection. To check the 3D position of each selected epitope in the protein structure, we used molecular homology modeling. Afterward, each selected epitope was evaluated by molecular docking to reference MHC-I or MHC-II allelic protein sequences. Taken together, the results obtained in this study showed a rational search for a putative immunogenic peptide of SARS-CoV-2 structural proteins that can improve vaccine development using in silico approaches. The epitopes selected represent the most conserved sequence of new coronavirus and may be used in a variety of vaccine development strategies since they are also presented in the described variants of SARS-CoV-2.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
